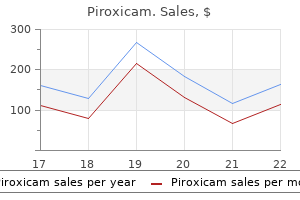
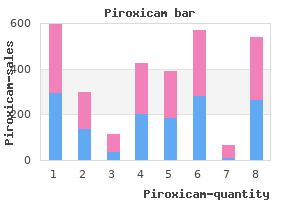
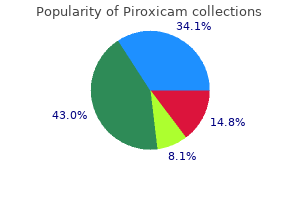
X




|
STUDENT DIGITAL NEWSLETTER ALAGAPPA INSTITUTIONS |

|
Sharon Phillips Andreoli, MD
Imprinted in hippocampal and cerebeller neurons rheumatoid arthritis cannabis buy piroxicam 20mg online, increases learning and long-term potentiation of synaptic transmission arthritis walk 2015 order 20 mg piroxicam fast delivery. Absence contributes to Angelman syndrome in humans arthritis diet tomatoes discount 20 mg piroxicam amex, which includes neonatal hyperactivity rheumatoid arthritis knuckles generic 20 mg piroxicam free shipping, opposite of the oppositely imprinted Prader-Willi syndrome migratory arthritis definition discount piroxicam 20mg online. Imprinted in fibroblasts arthritis medication pulled off market discount 20 mg piroxicam, lymphoblasts, and neural-precursor cells (Herzing et al. Expression limited to placenta; increases spongiotrophoblast growth at the expense of giant cells, which secrete placental lactogens (which actively solicit maternal investment; Haig 1993b). Giant cells penetrate farthest into maternal decidua; limitation on giant cells suggests limitation on maternal investment. Weakly imprinted only in extraembryonic tissues, biallelic in others, codes for a membrane protein, expressed in many tissues; no evidence on imprinted phenotype. Imprinted only in fetal tissues of mice and humans, biallelic in adults, encodes a voltage-sensitive potassium channel. Strongly imprinted in placenta (and liver) of mice and humans and weakly imprinted in other fetal and adult tissues; encodes a small cytoplasmic protein, restrains placental growth but without reducing fetal growth. Imprinted in mouse placenta, X-linked so probably imprinted as a result of general paternal X-inactivation (see Xist). Absence of gene increases size of early placenta but, at later stages, depresses fetal growth, probably due to placental pathology. Catalyzes the rate-limiting step in the synthesis of creatine, an important molecule in energy metabolism. Imprinted in placenta and yolk sac but not embryo; effect on offspring and maternal resources unknown (Sandell et al. Potent growth inhibitor: deleting maternal copy leads to placental and fetal overgrowth, especially liver but not brain, and offspring are 3040% larger at birth, independent of Igf2 pathway (Charalambous et al. Except where otherwise noted, all information (including almost every evaluation regarding kinship theory) is from Tycko and Morison (2002). When organized according to whether the genes are paternally active or maternally active and whether they affect growth positively or negatively, 10 strongly support the kinship theory, 8 weakly (at least 1 piece of evidence absent), and none is clearly opposed. In one case (Rasgrf1), the expected growth effect occurs after birth but before the end of weaning (Itier et al. In addition, some of the neutral cases are at least suggestive-for example, early growth effects not yet shown to be imprinted. In any case, the pattern is so strong as to be impervious to minor decisions regarding evaluation of individual cases (for detailed analysis of selected cases, see also Haig 2004b). Mice chimeras in which the added cells are androgenetic (double dose of paternal) are, as expected, usually larger than pure wildtype individuals, which in turn are larger than chimeras in which the added cells are parthenogenetic (Fundele et al. These differences tend to disappear after weaning, again as expected because there is little scope for internal genetic conflict over growth when the resources to support growth are supplied from outside the family. Humans are very unusual in the length of the period of parental investment-well past weaning and usually into young adulthood. Thus, important imprinted effects concerning parental investment may concern postweaning life. With this in mind, Haig and Wharton (2003) have recently reinterpreted the symptoms of Prader-Willi syndrome, which results from the absence of the paternal copy of a segment of chromosome 15. For the first few years of life, children suffer lassitude and low appetite, as expected from loss of paternally active genes during nursing; but then appetite becomes voracious and undifferentiated, that is, the children eat anything and even forage afield for new foods, traits that would tend to reduce cost to the mother. For a detailed analysis of imprinted effects on calcium metabolism in pregnancy, see Haig (2004a). Some genes may be imprinted as a pleiotropic effect of imprinting at a neighboring locus and some because in the past there was selection for a change in expression level, and the first appropriate mutation happened to work in a parent-specific manner. Once the imprinting machinery has evolved, we should expect non-kin effects to occur at least some fraction of the time. Evolution of the Imprinting Apparatus the Mechanisms of Imprinting Involve Methylation and Are Complex In order for maternal and paternal alleles to have different expression levels despite having the same nucleotide sequence, there must be some "epigenetic" difference between them. Methyl groups can be added or removed from particular C residues, and methylation states can be maintained or not through cell divisions, according to which enzymes are active. Among unimprinted genes, methylation is usually associated with gene silencing-so, for example, a gene required only in 1 tissue may be methylated and therefore silent in other tissues. Among imprinted genes, maternal and paternal alleles usually have different patterns of methylation; however, there is no consistent association between methylation and silencing. H19 and Snrpn show biallelic expression in methylation-deficient mice (indicating methylation is associated with silencing), but Igf2 and Igf2r show no expression (indicating methylation is associated with activation; Reik and Walter 2001a). Overall, it seems that for about half of imprinted genes, the methylated allele is the active one (Kaneko-Ishino et al. The removal occurs in the primordial germ cells of male and female fetuses and occurs as these cells are entering the developing gonad. It is not yet clear if the methyl groups are lost by active demethylation or passively (by failure to maintain methylation states through cell divisions). In males, methylation occurs in germline cells before meiosis (Reik and Walter 2001b). In females, de novo methylation occurs in oocytes, while they are arrested in meiosis I but still growing. Some genes are modified earlier than others-they are not all modified simultaneously-suggesting that different mechanisms may be used for different target genes (Obata and Kono 2002). Indeed, the association is so strong that the best way to find an imprinted gene is often to look near other imprinted ones. After implantation there is a complex series of secondary methylation imprints mediated by the control element, in both directions (Srivastava et al. As a consequence the promoter of the paternal H19 gene becomes methylated and is silenced, and a region upstream of the paternal Igf2 is methylated, which activates the gene. First, the paternal genome is actively demethylated just hours after fertilization, while the 2 parental genomes are still separate in the pronuclei. It might seem tempting to equate the germline methylation of a gene with the imprinting of that gene, and so Igf2r, for example, which is methylated in the female germline, would become a "maternally imprinted" gene. But there is a danger here if we conclude that the evolutionary innovation that led to parent-specific expression was some change in the female germline-in other words, that the ancestral state was no methylation in either germline. It is highly plausible, 107 Genomic Imprinting then, that the ancestral state was biallelic methylation, and the evolutionary innovation was the gain of this 6bp sequence that prevented methylation in the male germline. Once the maternal and paternal alleles have acquired their differential methylation, the final step in genomic imprinting is the use of those differences to affect gene expression. The first is the simplest and the most common: methylating a promoter in order to inactivate the gene. The second involves methylation of promoter regions of antisense transcripts that interact in cis to prevent expression of the corresponding sense transcripts. The third mechanism is regulation of genes by differential methylation of boundary elements within a CpG island. The methylated allele binds factors that block access of promoters to downstream enhancers. Finally, differential methylation may result in differential binding of silencing factors, which then repress the promoter in cis. With this mechanism methylation is associated with the active allele because it inactivates the silencers. Because CpG islands and clustered direct repeats attract methylation, it is perhaps not surprising that they are found close to the promoters of most imprinted genes (Reik and Walter 2001b). Likewise, how does imprinting work at Mash2, which continues to show maternal-only expression even in methylation-deficient mice (Kaneko-Ishino et al. For 6 genes (*) imprinting status is uncertain in humans, so the mouse status is given instead. Note there are only 8 unimprinted genes (central column) compared to 14 imprinted in the cluster, even though unimprinted genes are 100 times more frequent in the genome at large. Differential methylation of boundary elements within a CpG island: methylation prevents the binding of proteins (vertical disk) that block the access of upstream promoters to downstream enhancers. Methylation leads to differential binding of silencing factors that repress the promoter in cis. Genomic Imprinting genes are highly transcribed in order to counteract opposing genes and because only 1 allele is active. Why are maternal and paternal copies in imprinted regions often replicated asynchronously (Rand and Cedar 2003, Gribnau et al. And why do imprinted regions show higher rates of recombination in the male but not female germline (opposite the usual pattern; Paldi et al. Despite all these uncertainties, one thing that does seem clear is that the molecular mechanisms responsible for genomic imprinting are very complex. For the Igf2-H19 regions, some 30 different deletions, insertions, and transgenes have been characterized that reveal a complex regulatory system that we still do not understand in full (Arney 2003). The next section shows how this complexity is an expected feature of the internal tensions within the system, especially between the imprinting machinery and the imprinted genes. Conflict Between Different Components of the Imprinting Machinery In thinking about the evolution of imprinting in mammals, we can reasonably imagine that the ancestral state (before intimate mother-fetal interactions evolved) was equal expression of the 2 alleles, at a level that was optimal for the offspring. Then there are two possibilities for the origin of imprinting (Sleutels and Barlow 2002). First, there may have been preexisting differences in the methylation activities of the male and female germlines, and a gene evolved a cis-acting regulatory sequence that attracted methylation differentially in the 2 germlines, with the consequence that it was differentially expressed according to whether it was maternally or paternally inherited. In principle, a growth promoter like Igf2 might have evolved both "down-regulate-when-maternal" and "up-regulate-when-paternal" mutations (and vice versa for a growth suppressor like Igf2r). Under this scenario, the gene has evolved a facultative expression pattern, with no change in the methylation machinery itself. After a succession of such mutations, the final state is 1 silent allele and the opposite set at its optimal level of expression. Alternatively, there might initially have been no evolution of the gene itself or its cis-acting regulatory sequences, but rather evolution of the transacting imprinting machinery, starting initially with one that marks the target gene equally in the 2 germlines and evolves to mark it differentially. Rather than a gene evolving a facultative expression pattern, we imagine parents evolving to manipulate expression levels in their offspring. As an aside, each of these scenarios could include evolutionary changes in both male and female germlines. While we could say that a gene is maternally or paternally expressed, or methylated, it does not make sense to say that a gene is maternally or paternally imprinted-it is both. If both cis-acting targets and trans-acting Imprinters can evolve, they can coevolve. And this brings up a subtlety in the kinship theory of imprinting that may not at first be apparent. The conflict over kinship that underlies genomic imprinting does not just occur between maternal and paternal alleles of an individual, but also occurs within the imprinting process itself, that is, between trans-acting Imprinter loci and their cis-acting target: the two may "disagree" over whether an imprint should be applied (Burt and Trivers 1998a). This conflict arises because the target gene is always transmitted along with its own imprint, but an unlinked Imprinter gene is transmitted along with its imprinted target only half the time. For a maternally derived allele in a fetus, the fetus is as important as the mother (both contain a single copy of the gene); but for an Imprinter allele active in the female germline, the fetus is worth only half as much as the mother (because it has only a one-half chance of being in the fetus). Similar considerations apply in the male germline, and the general rule is that the target gene will be selected to make the fetus more demanding than will the imprinting apparatus (Box 4. Therefore, if the imprint is to silence a growth promoter or activate a growth suppressor, the imprinting machinery may be selected to apply the imprint, but the target sequence selected to resist it. In principle, this could lead to a coevolutionary chase through sequence space in which the target keeps changing to prevent being recognized, and the Imprinters keep evolving to recognize it. If the effect of the imprint is to silence a growth suppressor or activate a growth promoter, the target may be selected to acquire an imprint, but the imprinting machinery selected not to apply it. In this case the target might evolve to mimic other imprinted genes, and the imprinting apparatus therefore selected to make ever-finer discriminations. These conflicts between Imprinters and their targets must follow as surely as those between maternal and paternal alleles. There is another class of genes involved in imprinting, with yet another opinion on the optimal level of expression of a gene: the unimprinted offspring genes that maintain the imprint through development and read it at the time of expression. Even if the imprinting machinery is selected to apply an imprint and the target selected to acquire it, unimprinted offspring genes may be selected to erase the imprint, or ignore it. Thus, for, say, Igf2, there are 5 classes of genes that may affect fetal expression levels, each with its own opinion about the optimal level of expression. History of Conflict Reflected in the Imprinting Apparatus Thus the various classes of genes involved in imprinting are expected, from first principles, to evolve according to complex and contrarian selection pressures. We suggest that the unusual complexity of the imprinting apparatus is the result of a long history of constant adjustments and counteradjustments among the various components, as such complexity is unlikely to arise in a few mutational steps and seems unnecessary otherwise. More specifically, the fact that a particular C in the maternally derived Igf2r is methylated in the oocyte and the 2-cell embryo, unmethylated at the 4-cell stage, and then remethylated at the 8-cell stage is suggestive of a history of conflict (Moore and Reik 1996). The genome-wide demethylations that occur around this time may also represent attempts to erase unwanted imprints. This suggests that demethylation will not be seen in the absence of extended mother-fetal interactions-which is true of zebrafish (Macleod et al. In mice and humans, Igf2r and Grb10 show maternal-specific methylation, and in mice this is associated with maternal-specific expression, but in humans expression is typically biallelic, as if the methylation differences have no effect (Killian et al. Indeed there appear to be many regions of the genome in which maternal and paternal copies are differentially methylated, but there is no detectable difference in gene expression (de la Casa-Esperуn and Sapienza 2003). These thresholds differ for the different classes of genes involved in genomic imprinting, as shown in the figures in this box. The top figure covers mutations that benefit the fetus at a cost to the mother, while the bottom covers the reverse situation (labeling of lines is in inverse order to that above). Finally, for unimprinted progeny genes, the thresholds are the average of those for maternally and paternally imprinted genes. The top figure also gives the stopping rule for maternal investment for the different types of genes (continue to invest until the benefit:cost ratio falls below the specified level), whereas the bottom figure can be read as indicating how much more important the fetus is than the mother for the various genes. Note that for k = 1, all conflict is parent versus offspring, and that as k gets smaller, the conflict gradually becomes more maternal versus paternal.
Samples can therefore be collected from family members and stored for future analysis of disorders that are currently not amenable to molecular analysis cure arthritis with diet trusted piroxicam 20 mg. Tests are undertaken to identify conditions such as disorders of amino acids arthritis of fingers and toes cheap piroxicam 20 mg fast delivery, organic acids and mucopolysaccharides arthritis in fingers images purchase piroxicam 20 mg fast delivery, lysosomal and lipid storage diseases arthritis in toes buy piroxicam 20 mg lowest price, and Figure 1 tylenol arthritis pain gel tabs purchase 20mg piroxicam visa. Tests for other metabolites or enzymes are performed when a diagnosis of a specific disorder is being considered arthritis in dogs meds effective piroxicam 20mg. In some cases the register functions as a reference list of cases for diagnostic information, but generally the system is used to facilitate patient management. Less often there is an attempt to actively ascertain all affected cases within a given population. To function effectively most registers contain information about relatives at risk as well as affected individuals and may contain information from genetic test results. This is important for children at risk who may not need counselling and investigation for many years. A unique aspect of a family based genetic register is that it includes clinically unaffected individuals who may require continued surveillance and enables continued contact with couples at risk of transmitting disorders to their children. Disorders suited to a register approach include dominant disorders with late onset such as Huntington disease and myotonic dystrophy where pre-symptomatic diagnosis may be requested by some family members or health surveillance is needed by affected individuals; X linked disorders such as Duchenne and Becker muscular dystrophy where carrier testing is offered to female relatives, and chromosomal translocations where relatives benefit from carrier testing. Registers can also provide data on the incidence and natural course of disease as well as being used to monitor the provision and effectiveness of services. Genetic register information is held on computer and is subject to the Data Protection Act. No one has his/her details included on a register without giving informed consent. Throughout, the family requires support in adjusting to the implications of genetic disease and the consequent decisions that may have to be made. History taking Diagnosis of genetic disorders is based on taking an accurate history and performing clinical examination, as in any other branch of medicine. The history and examination will focus on aspects relevant to the presenting complaint. When a child presents with birth defects, for example, information needs to be gathered concerning parental age, maternal health, pregnancy complications, exposure to potential teratogens, fetal growth and movement, prenatal ultrasound scan findings, mode of delivery and previous pregnancy outcomes. Information regarding similar or associated abnormalities present in other family members should also be sought. In conditions with onset in adult life, the age at onset, mode of presentation and course of the disease in affected relatives should be documented, together with the ages reached by unaffected relatives. Detailed examination of children with birth defects or dysmorphic syndromes is crucial in attempting to reach a diagnosis. A careful search should be made for both minor and major congenital abnormalities. Measurements of height, weight and head circumference are important and standard growth charts and tables are available for a number of specific conditions, such as Down syndrome, Marfan syndrome and achondroplasia. Other measurements, including those of body proportion and facial parameters may be appropriate and examination findings are often best documented by clinical photography. In some cases, clinical geneticists will need to rely on the clinical findings of other specialists such as ophthalmologists, neurologists and cardiologists to complete the clinical evaluation of the patient. The person attending the clinic may not be affected, but may be concerned to know whether he or she might develop a particular disorder or transmit it to any future children. In such cases, the diagnosis in the affected relative needs to be clarified, either by examination or by review of relevant hospital records (with appropriate consent). Apparently unaffected relatives should be examined carefully for minor or early manifestations of a condition to avoid inappropriate reassurance. In myotonic dystrophy, for example, myotonia of grip and mild weakness of facial muscles, sterno-mastoids and distal muscles may be demonstrated in asymptomatic young adults and indicate that they are affected. Subjects who may show signs of a late onset disorder should be examined before any predictive genetic tests are done, so that the expectation of the likely result is realistic. Some young adults who request predictive tests to reassure themselves that they are not affected may not wish to proceed with definitive tests if they are told that their clinical examination is not entirely normal. A search for associated anomalies in children with chromosomal disorders often includes cranial, cardiac and renal imaging along with tests for other specific components of the particular syndrome, such as immune deficiency. In some genetic disorders affected individuals may require regular investigations to detect disease-associated complications, such as cardiac arrhythmias and reduced lung function in myotonic dystrophy. Screening for disease complications in asymptomatic relatives at risk of a genetic disorder may also be appropriate, for example, 24-hour urine catecholamine estimation and abdominal scans for individuals at risk of von HippelLindau disease. Family pedigrees are drawn up and relevant medical information on relatives sought. It is important to record full names and dates of birth of relatives on the pedigree, so that appropriate hospital records can be obtained if necessary. Specific questions should be asked about abortions, stillbirth, infant death, multiple marriages and consanguinity as this information may not always be volunteered. When a pedigree is drawn, it is usually easiest to start with the person seeking advice (the consultand). Details of first degree relatives (parents, siblings and children) and then second degree relatives (grandparents, aunts, uncles, nieces and nephews) are added. If the consultand has a partner, a similar pedigree is constructed for his or her side of the family. The affected person (proband) through whom the family has been ascertained is usually indicated by an arrow. Confirmation of a clinical diagnosis may identify a defined mode of inheritance for some conditions. In others, similar phenotypes may be due to different underlying mechanisms, for example, limb girdle muscular dystrophy may follow dominant or recessive inheritance and the pedigree may give clues as to which mechanism is more likely. In cases where no clinical diagnosis can be reached, information on genetic risk can be given if the pedigree clearly indicates a particular mode of inheritance. However, when there is only a single affected individual in the family, recurrence risk is difficult to quantify if a clinical diagnosis cannot be reached. In many conditions, however, risks are expressed in terms of probabilities calculated from pedigree data or based on empirical risk figures. An important component of genetic counselling is explaining these risks to families in a manner that they can understand and use in decision making. Mendelian disorders due to mutant genes generally carry high risks of recurrence whereas chromosomal disorders generally have a low recurrence risk. For many common conditions there is no clearly defined pattern of inheritance 6 Figure 2. Similar phenotypes may be due to mutations at different loci (locus heterogeneity) or to different modes of inheritance. In autosomal recessive deafness there is considerable locus heterogeneity with over 30 different loci known to cause nonsyndromic severe congenital deafness. The risk to offspring of two affected parents will be 100% if their deafness is due to gene mutations at the same locus, but negligible if due to gene mutations at different loci. In some disorders, for example hereditary spastic paraplegia and retinitis pigmentosa, autosomal dominant, autosomal recessive and X linked recessive inheritance have been documented. Definite recurrence risks cannot be given if there is only one affected person in the family, since dominant and recessive forms cannot be distinguished clinically. Perception of risk is affected by the severity of the disorder, its prognosis and the availability of treatment or palliation. All these aspects need to be considered when information is given to individuals and families. The decisions that couples make about pregnancy are influenced partly by the risk of transmitting the disorder, and partly by its severity and the availability of prenatal diagnosis. A high risk of a mild or treatable disorder may be accepted, whereas a low risk of a severe disorder can have a greater impact on reproductive decisions. Conversely, where no prenatal diagnosis is possible, a high risk may be more acceptable for a lethal disorder than for one where prolonged survival with severe handicap is expected. In marriages between first cousins the chance of a child inheriting the same recessive gene from both parents that originated from one of the common grandparents is 1 in 64. A different recessive gene may similarly be transmitted from the other common grandparent, so that the risk of homozygosity for a recessive disorder in the child is 1 in 32. Marriage between first cousins generally increases the risk of severe abnormality and mortality in offspring by 35% compared with that in the general population. Marriage between first and second degree relatives is almost universally illegal, although marriages between uncles and nieces occur in some Asian countries. Marriage between third degree relatives (between cousins or half uncles and nieces) is more common and permitted by law in many countries. The offspring of incestuous relationships are at high risk of severe abnormality, mental retardation and childhood death. Only about half of the children born to couples who are first degree relatives are normal and this has important implications for decisions about termination of pregnancy or subsequent adoption. Uncleniece Half siblings 1/4 Double first cousins Third: First cousins 1/8 Half-uncleniece Fourth: First cousins once removed 1/16 Fifth: Second cousins 1/32 Figure 2. The process aims to help the individual or family to: understand: Genetic counselling has been defined as a communication process with both educative and psychotherapeutic aims. While genetic counselling must be based on accurate diagnosis and risk assessment, its use by patients and families will depend upon the way in which the information is given and its psychosocial impact addressed. The ultimate aim of genetic counselling is to help families at increased genetic risk to live and reproduce as normally as possible. While genetic counselling is a comprehensive activity, the particular focus will depend upon the family situation. A pregnant couple at high genetic risk may need to make urgent decisions concerning prenatal diagnosis; parents of a newly diagnosed child with a rare genetic disorder may be desperate for further prognostic information, while still coming to terms with the diagnosis; a young adult at risk of a late onset degenerative disorder may be well informed about the condition, but require ongoing discussions about whether to go ahead with a presymptomatic test; and a teenage girl, whose brother has been affected with an X linked disorder, may be apprehensive to learn about the implication for her future children, and unsure how to discuss this with her boyfriend. Adapted from American Society of Human Genetics, 1975 Psychosocial issues the psychosocial impact of a genetic diagnosis for affected individuals and their families cannot be over emphasised. The diagnosis of any significant medical condition in a child or adult may have psychological, financial and social implications, but if the condition has a genetic basis a number of additional issues arise. These include guilt and blame, the impact on future reproductive decisions and the genetic implications to the extended family. Parents very often express guilt at having transmitted a genetic disorder to their children, even when they had no previous knowledge of the risk. On the other hand, parents may also feel guilty for having taken the decision to terminate an affected pregnancy. Although in most situations the person expressing guilt will have played no objective causal role, it is important to allow him or her to express these concerns and for the counsellor to reinforce that this is a normal human reaction to the predicament. Although parents often fear that their children will blame them for their adverse genetic inheritance, in practice this happens infrequently and usually only when the parents have knowingly withheld information about the genetic risk. Some couples may be faced with a perplexing range of options including different methods of prenatal diagnosis and the use of assisted reproductive technologies. For others the only available option will be to choose between taking the risk of having an affected child and remaining childless. Couples may need to reconsider these choices on repeated occasions during their reproductive years. Most couples are able to make reproductive choices and this is facilitated through access to full information and counselling. Decision making may be more difficult in particular circumstances, including marital disagreement, religious or cultural conflict, and situations where the prognosis for an affected child is uncertain. For many genetic disorders with variable severity, although prenatal diagnosis can be offered, the clinical prognosis for the fetus cannot be predicted. When considering reproductive decisions, it can also be difficult for a couple to reconcile their love for an affected child or family member, with a desire to prevent the birth of a further affected child. For example, the parents of a boy just diagnosed with Duchenne muscular dystrophy will not only be coming to terms with his anticipated physical deterioration, but may have concerns that a younger son could be affected and that daughters could be carriers. This is likely to be distressing even when family relationships are intact, but will be further complicated in families where relationships are less good. Family support can be very important for people coping with the impact of a genetic disorder. When there are already several affected and carrier individuals in a family, the source of support from other family members can be compromised. They may also be hesitant to discuss decisions about predictive or prenatal testing with relatives who may have made different choices themselves. The need for an independent friend or counsellor in these situations is increased. A genetic disorder may lead to reproductive loss or death of a close family member. This is sometimes coordinated through regional family genetic register services, or may be requested by family members at important life events including pregnancy, onset of symptoms, or the death of an affected family member. In addition to the value of contact with other families who have personal experience of the condition, several groups now offer the help of professional care advisors. The extent of the counselling and the issues to be addressed will depend upon the type of test being offered, which may be diagnostic, presymptomatic, carrier or prenatal testing. It is therefore the responsibility of the clinician offering the test to inform the patient (or the parents, if a child is being tested) before the test is undertaken, that the results may have genetic as well as clinical implications. Confirming the diagnosis of a genetic disorder in a child, for example, may indicate that younger siblings are also at risk of developing the disorder. For late onset conditions such as Huntington disease, it is crucial that samples sent for diagnostic testing are from patients already symptomatic, as there are stringent counselling protocols for presymptomatic testing (see below). Presymptomatic testing Genetic testing in some late onset autosomal dominant disorders can be used to predict the future health of a well individual, sometimes many decades in advance of onset of symptoms. For some conditions, such as Huntington disease, having this knowledge does not currently alter medical management or prognosis, whereas for others, such as familial breast cancer, there are preventative options available. For adult onset disorders, testing is usually offered to individuals above the age of 18. For conditions where symptoms or preventative options occur in late childhood, such as familial adenomatous polyposis, children are involved in the testing decision.
Purchase piroxicam 20 mg mastercard. Rheumatoid Arthritis TREATMENT in WINTER | Ayurvedic Home Remedies in Telugu by Dr. Murali Manohar.
A bronchoscopy (with biopsy) was performed that identified squamous cell carcinoma arthritis nursing diagnosis 20 mg piroxicam. Design a specific chemotherapeutic regimen to treat this patient arthritis flare up in neck cheap piroxicam 20mg without a prescription, and explain why you chose this regimen rheumatoid arthritis joint pain effective 20 mg piroxicam. What additional measures should be taken to ensure the tolerability of the regimen and to prevent adverse effects? What additional laboratory and clinical information is needed before administration of the chemotherapy? What clinical and laboratory parameters are necessary to evaluate the therapy for achievement of the desired therapeutic outcome and the occurrence of adverse effects? What information should be provided to the patient to optimize therapy and minimize adverse effects? At one point arthritis of the jaw generic piroxicam 20 mg online, the patient presented with a serum calcium level of 12 mg/dL and an albumin of 1 arthritis diet what not to eat cheap piroxicam 20mg mastercard. Review clinically important drug interactions for cancer patients started on phenytoin arthritis fat fingers order 20 mg piroxicam. Describe the treatment goals associated with early and advanced stages of colon cancer. Design an appropriate chemotherapy regimen for colon cancer based on patient-specific data. Formulate a monitoring plan for a patient receiving a prescribed chemotherapy regimen for colon cancer based on patient-specific information. Recommend alterations in a drug therapy plan for a patient with colon cancer based on patient-specific information. Educate patients on the anticipated side effects of irinotecan, capecitabine, oxaliplatin, and epidermal growth factor receptor inhibitors. American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. Chemotherapy plus radiotherapy compared with radiotherapy alone in the treatment of locally advanced, unresectable, non-small-cell lung cancer. American Society of Clinical Oncology guideline for antiemetics in oncology: update 2006. He underwent surgical resection and lymphadenectomy for an obstructing lesion in the transverse colon. The pathology revealed poorly differentiated adenocarcinoma with increased vascular invasion through the entire thickness of the bowel wall. Sixteen days later, he presented to an outside hospital with complaints of abdominal pain, nausea, vomiting, and constipation. After five cycles of chemotherapy, he achieved a partial response in his liver metastases. His paternal grandfather died in his 60s from colon cancer; he is aware of no other family history of malignancy. He denies fever, headaches, shortness of breath, cough, nausea, vomiting, or diarrhea. He has been experiencing difficulty moving his bowels, but there is no pain or blood with passage of stool. What additional drug treatment interventions should be considered for this patient? What acute adverse effects are anticipated with the chemotherapy regimen, and what parameters should be monitored? What pharmacologic measures can be instituted to prevent or manage the acute toxicities associated with the chemotherapy regimen? What are the potential late-onset toxicities of the chemotherapy regimen, and how can they be detected and prevented? What information should you provide to the patient to enhance compliance, ensure successful therapy, and minimize adverse effects? His transdermal fentanyl dose was increased to 150 mcg/h every 72 hours, and oral immediate-release morphine sulfate, every 4 hours as needed for pain, was started. A scheduled regimen of docusate sodium, 100 mg orally plus 2 senna tablets daily, maintained a regular pattern of bowel movements. Eight months after starting chemotherapy, he presented with worsening pain, fatigue, and new-onset dyspnea. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. How should a patient who develops a thrombotic event during bevacizumab therapy be managed? After considering various treatment options with his oncologist, he agreed to treatment with an epidermal growth factor receptor inhibitor. Describe the standard initial treatment options for androgendependent metastatic prostate cancer. Recommend a pharmacotherapeutic plan for patients with androgen-independent metastatic prostate cancer. Counsel patients regarding the toxicities associated with the pharmacologic agents used in prostate cancer treatment. Develop patient-specific education materials regarding management of cutaneous toxicities of agents used in colon cancer treatment. The optimal combination, sequence, and treatment duration are unknown, but the ability of an individual to receive all active agents during the course of the disease is associated with the greatest likelihood of increasing overall survival. Family history of cancer: father, lung: diagnosed age 71, died age 73; mother, breast: died at age 93. He has a paternal aunt and paternal grandmother who both were diagnosed with unspecified malignancies. Neck/Lymph Nodes No cervical or supraclavicular adenopathy Lungs/Thorax Lungs are clear in all fields. Multiple small, external iliac lymph nodes are present, predominantly on the left. Patient has metastatic androgen-dependent hormone-sensitive disease and is here for consideration of initial treatment options. What signs, symptoms, and other information are consistent with metastatic prostate cancer in this case? Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. Docetaxel and estramustine compared with mitoxantrone and prednisone for refractory prostate cancer. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. How should the therapy you recommended be monitored for efficacy and adverse effects? He is complaining of increased pain in his pelvis and more bone pain in his ribs and back over the last 2 months, although he is still able to participate in church social activities and play golf on the weekends. His bone scan shows numerous intense foci in the skull, scapulae, spine, and femurs. What pharmacotherapeutic options are available to the patient for his progressive androgen-independent metastatic prostate cancer? Locate information resources that are available to prostate cancer patients and their families. Physical examination findings were significant for decreased breath sounds (worse on the left side than the right) and enlarged, painless supraclavicular lymph nodes on the left side. Chest x-ray revealed a large heterogeneous mass at the apex of the left lung also involving the mediastinum. Pathology revealed cells consistent with lymphoma, but definitive diagnosis could not be made. The oncologist on call was consulted, and it was recommended for him to follow up as an outpatient for further evaluation and treatment recommendations. He states that he does not eat many vegetables, as he must save room for "the good stuff" on the buffet. In addition, he describes an unexplained weight loss of approximately 25 pounds over the last 3 months. He states that he occasionally has some dyspnea on exertion, but he is able to carry out activities of daily living without limitations. Genit/Rect Normal male genitalia Ext Without edema, warm to the touch; pulses palpable bilaterally Neuro Symmetric cranial nerve function. Balance and coordination of the upper extremities are intact, with no evidence of tremor. Lymph Node Survey the lymph node survey is negative for any palpable peripheral nodes in the preauricular, postauricular, cervical, supraclavicular, infraclavicular, or axillary areas. What pharmacologic measures should be instituted to treat or prevent the acute toxicities associated with the chemotherapy regimen? What are potential late complications of the chemotherapy regimen, and how can they be detected and prevented? He was initiated on darbepoetin, 500 mcg every 3 weeks, and ferrous sulfate, 325 mg 3 times daily. The darbepoetin dose was subsequently reduced to 300 mcg every 3 weeks due to hemoglobin increase above 11 g/dL. The patient was stabilized, and appropriate modifications were made to his home medications for treatment of new-onset heart failure. Imaging studies were also performed while he was in the hospital to evaluate his lymphoma. Explain what system of staging was used and how his stage of disease was determined. What laboratory and clinical features does this patient have that may affect his prognosis? What nonanthracycline-containing regimens are available for this patient to complete his chemotherapy course? He and his wife decided that they did not want to undergo any more chemotherapy because he is currently in remission after just four cycles of chemotherapy. Eighteen months later, the patient returns to the oncologist office after being diagnosed with relapsed lymphoma during a hospital admission for worsening dyspnea. What therapeutic options are available for the treatment of relapsed diffuse large B-cell lymphomas? If the patient experienced tumor lysis syndrome, what options are there for treating the hyperuricemia? Numerous clinical trials are under way evaluating the role of hematopoietic stem cell transplantation, different chemotherapy combinations, and new monoclonal antibodies with different targets. What acute adverse effects are associated with the chemotherapy regimen, and what parameters should be monitored? A proposal for a simple staging system for intermediate grade lymphoma and immunoblastic lymphoma based on the "tumor score. She states that the nodes in her neck "come and go" but are always somewhat swollen; she relates this to a sore throat. The node in the axilla has been consistently swollen and painful during this time. She also complains of new-onset fatigue and general low back pain for the last 45 months. She denies any shortness of breath but has experienced fever, night sweats, and weight loss for the past few months. An ultrasound of the neck and axilla showed a number of lymph nodes ranging from 1. She began smoking at the age of 18 and smoked socially, two cigarettes a week, until the age of 24. There is involvement of the superior middle mediastinum and hilar nodal chains within the chest. There are multiple, enlarged para-aortic and right iliac lymph nodes in the abdomen and pelvis. Diffuse increased uptake within the bone marrow; it is unclear whether this is lymphoma or hyperplasia. She received day 1 of her first cycle of chemotherapy and experienced acute nausea and vomiting. At that time, the patient restarted her chemotherapy regimen and received pegfilgrastim 24 hours after her chemotherapy to maintain her current dose of chemotherapy without dose reduction and dose delay. As a result of pegfilgrastim, the patient experienced severe bone pain that was relieved by oxycodone/ acetaminophen. Restaging showed the patient had a complete response and on subsequent follow-up is in remission. What is the antiemetic regimen of choice to prevent acute nausea and vomiting for highly emetogenic chemotherapy? Explain what system of staging was used and how her stage of disease was determined. Recommend a pharmacotherapeutic plan for the chemotherapeutic treatment of newly diagnosed and relapsed ovarian cancer. Describe the uses and potential pharmacologic advantages of intraperitoneal therapy for treatment of ovarian cancer. Recognize the dose-limiting and most common toxicities associated with the chemotherapeutic agents used in the treatment of ovarian cancer. Exploratory laparotomy revealed a 20 Ч 10 cm mass near the left ovary and positive microscopic disease in the omentum. Tumor biopsies from the ovary were positive for epithelial ovarian cancer with serous histology. Biopsies of the pancreas and abdominal/pelvic mass were positive for recurrent epithelial ovarian cancer. Erving is now admitted for her first cycle of chemotherapy for her relapsed ovarian cancer.
The 1 collagen gene encodes the two blue polypeptide chains arthritis in dogs front leg treatment 20 mg piroxicam with amex, and the 2 procollagen gene encodes the third (red) chain can you cure arthritis in the knee discount piroxicam 20 mg fast delivery. The procollagen triple helix is shortened before it becomes functional arthritis knuckles 20 mg piroxicam mastercard, forming the fibrils and networks that comprise much of the human body arthritis cyst buy discount piroxicam 20 mg on-line. Detection of mutations that cause Marfan syndrome before symptoms arise can be lifesaving arthritis medication options buy 20mg piroxicam otc, because frequent ultrasound exams can detect aortic weakening early enough to patch the vessel before it bursts arthritis and diet mayo clinic piroxicam 20 mg fast delivery. For one gene, different mutations may cause differing degrees or different subsets of symptoms of one syndrome. Yet different mutations in the beta globin gene cause the clinically distinct sickle cell disease and beta thalassemia, two different disorders, although Figure 12. A mutation that blocks trimming of procollagen chains to produce collagen causes the stretchy skin of EhlersDanlos syndrome type I. Deletion eliminates dystrophin, which normally binds to inner face of muscle cell plasma membranes, maintaining cellular integrity. Extra bases in the gene add amino acids to the protein product, which impairs certain transcription factors and proteasomes. Defect in protein that normally suppresses activity of a gene that causes cell division. Lamin A mutations cause the rapid-aging disorder Hutchinson-Gilford progeria syndrome (see figure 3. Lamin A proteins form a network beneath the inner nuclear membrane that interacts with chromatin. For example, two healthy people of normal height may have a child with achondroplasia, an autosomal dominant form of dwarfism. His siblings have no higher risk of inheriting the condition than anyone in the general population, but each of his children will face a 50 percent chance of inheriting it. One cause of spontaneous mutation stems from the chemical tendency of free nitrogenous bases to exist in two slightly different structures, called tautomers. Mutations add, delete, or rearrange genetic material in a germline cell or somatic cell. In sickle cell disease, a mutation causes hemoglobin to crystallize in a low-oxygen environment, bending red blood cells into sickle shapes and impairing circulation. In beta thalassemia, beta globin is absent or scarce, depleting hemoglobin molecules. Mutations in a gene may cause either different versions of the same disease or distinct illnesses. Each human gene has about a 1 in 100,000 chance of mutating and each of us probably carries a few new spontaneously mutated genes. This is possible because a new dominant mutation is detectable simply by observing the phenotype. In contrast, a new recessive mutation would not be obvious until two heterozygotes produced a homozygous recessive offspring with a noticeable phenotype. The spontaneous mutation rate for autosomal genes can be estimated using the formula: number of de novo cases/2X, where X is the number of individuals examined. The denominator has a factor of 2 to account for the nonmutated homologous chromosome. Spontaneous mutation rates in human genes are difficult to assess because our generation time is long-usually 20 to 30 years. In bacteria, a new generation arises every half hour or so, and mutation is therefore much more frequent. If a replication fork encounters a base in its unstable form, a mismatched base pair can result. Gene Mutation © the McGraw-Hill Companies, 2010 223 Mutational Hot Spots In some genes mutations are more likely to occur in regions called hot spots, where sequences are repetitive. It is as if the molecules that guide and carry out replication become "confused" by short repeated sequences, much as an editor scanning a manuscript might miss the spelling errors in the words "happpiness" and "bananana" (figure 12. Put another way, the sequence on one strand is the reverse of the sequence on the complementary strand. Palindromes probably increase the spontaneous mutation rate by disturbing replication. A person who does not have the disorder has four genes that specify alpha globin chains, two next to each other on each chromosome 16. Homologs with repeated genes can misalign during meiosis when the first sequence on one chromosome lies opposite the second sequence on the homolog. Crossing over can result in a sperm or oocyte that has one or three alpha globin genes instead of the normal two (figure 12. Fertilization with a normal gamete then results in a zygote with one extra or one missing alpha globin gene. At least three dozen conditions result from this unequal crossing over, including colorblindness (see Reading 6. A person with only three alpha globin genes produces enough hemoglobin, and is a healthy carrier. Individuals with only two copies of the gene are mildly anemic and tire easily, and a person with a single alpha globin gene is severely anemic. The repeated alpha globin genes are prone to mutation by mispairing during meiosis. Gene Mutation © the McGraw-Hill Companies, 2010 of genetic variants for experiments, researchers make mutants. Geneticists use mutagens on model organisms to infer normal gene functions, yielding many collections and insights into human health. This occurs from workplace contact before the danger is known; from industrial accidents; from medical treatments such as chemotherapy and radiation; and from exposure to weapons that emit radiation. An environmental disaster that released mutagenic radiation was a steam explosion at a nuclear reactor in the former Soviet Union on April 25, 1986. The reactor had been undergoing a test, its safety systems temporarily disabled, when it overloaded and rapidly flared out of control. Twenty-eight people died of acute radiation exposure in the days following the explosion. Evidence of a mutagenic effect is the increased rate of thyroid cancer among children who were living in nearby Belarus. The thyroid glands of young people soak up iodine, which in a radioactive form bathed Belarus in the days after the explosion. Analysis of radiation exposure in their teeth is being used to assess whether cancer risk rises with degree of exposure. Such a mutation was twice as likely to occur in exposed families as in families living elsewhere. Several other mutagenic chemicals alter base pairs, so that an A-T replaces a G-C, or vice versa. One version of the test uses a strain of Salmonella that cannot grow when the amino acid histidine is absent from its medium. If exposure to a substance enables bacteria to grow on the deficient medium, then a gene has mutated that allows it to do so. In another variation of the Ames test, researchers exposed human connective tissue cells growing in culture to liquefied cigarette smoke. This is an especially damaging insult because broken chromosomes can join with each other in different ways that can activate cancer-causing genes. A limitation of using a mutagen is that 2-amino 5-nitrophenol Hair dye components it cannot cause a specific mutation. In con2,4-diaminoanisole trast, a technique called site-directed mutagenesis changes a gene in a desired way. A gene is 2,5-diaminoanisole mass-produced, but it includes an intentionally 2,4-diaminotoluene substituted base, just as an error in a manuscript is printed in every copy of a book. Site-directed p-phenylenediamine mutagenesis is faster and more precise than Furylfuramide Food additive waiting for nature or a mutagen to produce a useful variant. It also makes it possible to study Nitrosamines Pesticides, herbicides, cigarette smoke lethal mutations that can theoretically exist, but Proflavine Antiseptic in veterinary medicine never do because they are so drastic that development does not proceed very far. Gene Mutation © the McGraw-Hill Companies, 2010 225 sequences are too low to provide useful information on the effects of radiation exposure, so investigators track minisatellites as a sensitive test of change. Researchers learned of a new type of mutation from a young man conceived within a week of the Chernobyl accident, near the disaster site. Researchers indeed found a mutation in the gene on chromosome 7 known to cause the syndrome-a 72-base insertion that causes a "stop" codon to form, shortening the encoded protein. Contributions from medical X rays and occupational radiation hazards are comparatively minor (table 12. Job sites with increased radiation exposure include weapons facilities, research laboratories, health care facilities, nuclear power plants, and certain manufacturing plants (figure 12. Radiation exposure is measured in units called millirems; the average annual exposure in the northern hemisphere is 360 millirems. Most of the potentially mutagenic radiation we are exposed to is of the ionizing type, which means that it has sufficient energy to remove electrons from atoms. Unstable atoms that emit ionizing radiation both exist naturally and are made by humans. Alpha radiation is the least energetic and most short-lived, and the skin absorbs most of it. Beta radiation can penetrate the body farther, and emitters include tritium (a form of hydrogen), carbon-14, and strontium-70. Both alpha and beta rays tend not to harm health, although they can do damage if inhaled or eaten. Plutonium and cesium isotopes used in weapons emit gamma rays, and this form of radiation is used to kill cancer cells. X rays are the major source of exposure to human-made radiation, and they are not a form of ionizing radiation. They have less energy and do not penetrate the body to the extent that gamma rays do. Mutations in oncogenes or tumor suppressor genes, discussed in chapter 18, can cause cancer. Exposing cells to radiation and then culturing them causes a genome-wide destabilization, so that mutations may occur even after the cell has divided a few times. Cell culture studies have also identified a "bystander effect," when radiation harms cells not directly exposed. Evaluating the risk that a specific chemical exposure will cause a mutation is very difficult, largely because people vary greatly in inherited susceptibilities, and are exposed to many chemicals. The risk that exposure to a certain chemical will cause a mutation is often less than the natural variability in susceptibility within a population, making it nearly impossible to track the true source and mechanism of any mutational event. Genetic tests can be used to determine specific inherited risks for specific employees who might encounter a mutagen in the workplace. Exposure to radiation can cause mutation, whether from an unnatural source, such as this nuclear power plant, or a natural source, such as the sunlight that enables the field of rapeseed in the foreground to grow. Spontaneous mutations are more frequent in viruses and microorganisms because they reproduce often. Researchers use mutagens to more quickly obtain mutants, which reveal normal gene function. It is a transition if a purine replaces a purine (A to G or G to A) or a pyrimidine replaces a pyrimidine (C to T or T to C). It is a transversion if a purine replaces a pyrimidine or vice versa (A or G to T or C). A point mutation can have any of several consequences-or it may have no obvious effect at all on the phenotype, acting as a silent mutation. Missense and Nonsense Mutations A point mutation that changes a codon that normally specifies a particular amino acid into one that codes for a different amino 6. Mutagens are encountered in the natural environment acid is called a missense mutation. The resulting protein is longer than + Gln Pro O normal, because translation continues Leu Ser through what is normally a stop codon. The mutation is in the proresults from a nonsense mutation, in which a stop codon replaces a tryptophan codon. Gene Mutation © the McGraw-Hill Companies, 2010 227 sensation and involuntary responses. Current clinical trials are examining the ability of several natural compounds to restore normal processing of the slows transcription, and dystrophin protein is scarce. The other 85 percent of individuals who have Becker muscular dystrophy have shortened proteins, not a defiDeletions and Insertions ciency of normal-length proteins. Adding or deleting a number duction is to disrupt the trimming of long precursor molecules. An exon is usually "readable" (has no Splice Site Mutations stop codons) in only one of its three possible reading frames. Many common inherited disorders result from deletions, severe cystic fibrosis, a missense mutation alters an intron site including male infertility caused by tiny deletions in the Y so that it is not removed. The mutation creates an intron splicing site where there of an enzyme that normally breaks down glycolipids in lysoshould not be one, and an entire exon is "skipped" when the somes. The insertion is usually adjacent or close results from exon skipping in the gene encoding an enzyme to the original sequence, like a typographical error repeatnecessary for the survival of certain neurons that control ing a word word. This is a rare genetic disorder that affects the autonomic and peripheral nervous systems. She appeared to be healthy at birth, but she began to decline rapidly by 9 months.
References